Introduction
Physical activity is one of the most important elements in prevention of civilization diseases and diseases connected with the aging process. The advantages of being physically active are much more significant than risks connected with it (Jansson et al., 2015). Physical activity, with adequate volume and intensity, is one of the cheapest and most universal medicine with only small side effects. It can be treated as “home pharmacy” that has been used by many generations (Grzywacz and Jaroń, 2020; Tuka et al., 2017).
Regular physical activity has shown to protect against numerous kinds of cancers in different populations and in various settings. It is well documented that a higher level of physical activity decreases the risk of colorectal, breast, and endometrial cancer (Friedenreich et al., 2010; Kyu et al., 2013). There are new data suggesting the crucial role of exercise in prevention of other types of cancer. A current pooled analysis of 1,44 million individuals allows to notice the extended protective effects connected with head and neck, esophageal, lung, kidney, blood, and bladder cancers without any dependence on the body mass index (Moore et al., 2016). Hence, it can be concluded that the influence of physical inactivity is of high importance in different cancers. Data from 2013 show that nearly 7% of colorectal and breast cancer cases worldwide occur because of physical inactivity, what created a total healthcare cost of $5.2 billion for only those two cancers (Ding et al., 2016). The biological mechanisms influencing cancer risk in connection with physical inactivity has been analyzed for healthy populations in recent randomized controlled trials (Campbell et al., 2012; Friedenreich et al., 2014; Monninkho et al., 2007; Van Gemert et al., 2013). DNA methylation is one of the mechanisms recently identified, but still poorly understood in relation to physical inactivity and carcinogenesis prevention. The process of aging and exposure to carcinogenic agents can dysregulate patterns of DNA methylation, what can provoke genomic instability and improper gene expression (Berdasco and Esteller, 2010; Boyne et al., 2018; Jones et al., 2015; Kochmanski et al., 2017).
Exercise and dietary interventions, which are environmental stimuli, can be elements modifying DNA methylome at a global and gene-specific level (Alegría-Torres et al., 2011). The process of hypomethylation after exercise was observed for both skeletal muscles (Nitert et al., 2012; Rowlands et al., 2014; Seaborne et al., 2018) and blood leukocytes (Denham et al., 2015, 2016; Dimauro et al., 2016). Hypomethylation was observed in skeletal muscles after exercise (Bajpeyi et al., 2017; Barrès et al., 2012; Lane et al., 2015; Seaborne et al., 2018), however, the only research considering DNA methylation in leukocytes in the same situation did not allow to detect any changes in DNA methylation (Robson-Ansley et al., 2014). Considering the small number of publications that discuss the impact of acute exercise on DNA methylation in leukocytes, the researchers suggest an epigenetic influence connected with remodeling of the leukocyte transcriptome (Büttner et al., 2007; Connolly et al., 2004; Gjevestad et al., 2015; Hunter et al., 2019).
Phenotype transmission and the occurrence of different diseases, both seem to be induced by epigenetic mechanisms, among them DNA methylation (Fraga et al., 2005; Gluckman et al., 2009; Ling et al., 2007; Ronn et al., 2008; Sandovici et al., 2011). The epigenetic pattern is mostly imprinted in early life stages, but its modulating potential connected with gene expression and environmental factors influencing phenotypic traits is still observed later in life (Rönn et al., 2013). Changes most often taking place in differentiated mammalian cells include DNA methylation, usually occurring in the context of CG dinucleotides (CpGs) and is connected with repression of genes (Bird, 2002). The stimuli which can provoke changes in epigenetic profiles can be divided into environmental, behavioral, psychological and pathological. What is also important the epigenetic changes are more common than mutations (Feinberg and Irizarry, 2010). Furthermore, genetic variation not connected with a phenotype could affect the number of variability of that phenotype through epigenetic mechanisms, among them DNA methylation. Regular exercise and the mechanism of its long-lasting influence are still not well understood, hence many projects concentrate on cellular and molecular changes in skeletal muscles. Current knowledge about DNA methylation in human skeletal muscles emphasizes changes occurring in the epigenetic pattern as an effect of long-term exercise (Rönn et al., 2013). Animal models are applied to analyze numerous physiological and behavioral phenotypes in the context of regulatory influence of DNA methylation. What is important, there are proves showing that it is involved in neuronal and brain development (Watson et al., 2015; Wilson and Sengoku, 2012; Spiers et al., 2015) In human studies, the process of unbalanced methylation has been observed among individuals with substance dependency, anxiety, depression, autism, schizophrenia, and bipolar disorder (Grzywacz et al., 2020; Renthal and Nestler, 2008; Schmitt et al., 2014; Spiers et al., 2015; Tuesta and Zang, 2014)
Since in our project the research group was composed of martial arts athletes, we selected the area of the gene connected with dopamine and dopaminergic transmission to be analyzed. Dopamine is the substance often considered an element responsible for making the so called “risky decisions”. Worth mentioning is also the “mesolimbic reward system” that mediates in reward psychopharmacology influenced by a physical effort and other factors. Nonetheless, in this case the brain region of the ventral tegmental area, where neurons of the dopaminergic system are situated, and the nucleus accumbens, which is functionally connected to it, are called the “pleasure center” and dopamine itself is called the “pleasure neurotransmitter”. Hence, when considering the above mentioned system, we can assume that it can be treated as one of the key determinants of starting and continuing athletic training. Thus, the element influencing taking up and continuing athletic training and possibility of sport success, is the interaction of genetically determined temperament and the environmentally determined character. It is also worth to emphasize additional, physiological reasons to begin studies analyzing the genetic mechanisms determining operations of the dopaminergic system in the context of physical effort and sport. Noticing the fact that dopamine, as a neurotransmitter, is also engaged in neural transmission, e.g. in the extrapyramidal system, one could argue that it could also be of fundamental importance to the level of motor coordination. Some studies on the animal model have noticed that in specific situations of dopamine stimulation deficit mammals become immobile (akinetic), and increased dopamine stimulation provokes greater mobility.
In the context of numerous studies and reports, in our project we tried to consider the methylation process in the group of athletes training regularly in comparison with control subjects that included healthy people, who were not regularly engaged in physical activity. The dopamine transporter DAT1 and methylation in the promoter region were chosen to be analyzed.
Methods
Participants
The research group included 100 athletes (mean age = 22.88, SD = 6.35), whereas the control group included 239 healthy male volunteers matched for age (mean age = 21.69, SD = 3.39). Both, the control and the research group, included individuals with Caucasian origin from the same region of Poland. The study was conducted among 100 Polish healthy (no prior history of substance dependency or psychosis) male combat athletes aged 22.88 ± 6.35 (MMA, n = 23; judo, n = 40; boxing, n = 5; karate, n = 15; kickboxing, n = 15; wrestling, n = 2). Various methods were used to obtain the samples, including targeting national teams and providing information to national coaching personnel and athletes attending training camps. All athletes and controls were Caucasian to reduce the possibility of racial gene skewing and to overcome any potential problems due to population stratification.
Methylation status assessment
DNA was extracted from peripheral blood leukocytes using a DNA isolation kit (A&A Biotechnology, Gdynia, Poland) as previously described and stored at -20ºC. Bisulfite modification of 250 ng DNA was performed using the EZ DNA Methylation Kit (Zymo Research, Orange, CA, USA), according to manufacturer's instructions. The methylation-specific PCR assay was carried out in a Mastercycler epgradient S (Eppendorf, Germany).
Primer oligonucleotides were obtained from Genomed.pl (Warsaw, Poland). Primer sequences were designed using methprimer (http://www.urogene.orgbin/methprimer/.cgi) The status of the DAT1 promoter (ENSG00000142319) was assessed by PCR using primers specific to a fragment of the gene, i.e., DATF: 5’-GGTTTTTGTTTTTTTTATTGTTGAG-3’; DATR: 5’-AAATCCCCTAAACCTAATCCC-3’. The PCR conditions in order to amplify the 447-bp fragment covering 33 CpG sites in DAT1 gene promoter were as follows: initial denaturation (94°C/5 min), followed by 35 cycles (94°C/61°C/72°C, 25 s each step) with final elongation at 72°C for 5 min. The concentration of magnesium chloride ions was 2.5 mM. After the amplification assay, PCR products were subjected to sequencing as previously described (Kochmanski et al., 2017). Briefly, samples were verified by sequencing using the BigDye v3.1 kit (Applied Biosystems, Darmstadt, Germany) and separation by ethanol extraction using the ABI Prism 3130XL (Applied Biosystems, Darmstadt, Germany) in a 36 cm capillary in a POP7 polymer, using the reverse primer.
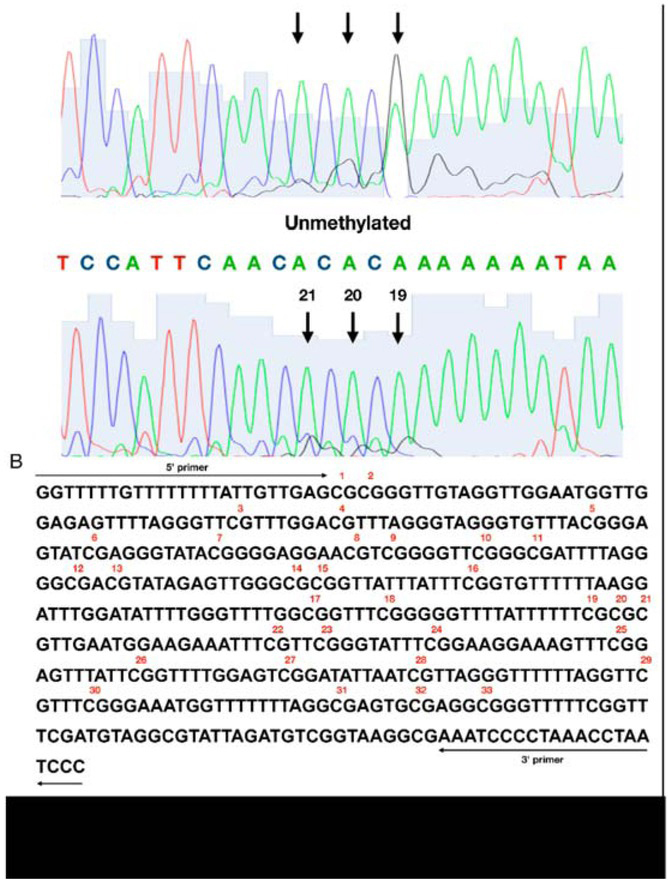
Sequencing chromatograms were analyzed using 4peaks software (Mek & Tosj, Amsterdam, The Netherlands). Methylation of cytosine was considered positive, when the G/A+G ratio reached at least 20% of a total signal (Figure 1).
Figure 1
An assessment of a methylation status of individual CpG sites in the DAT1 promoter. (A) The representative result of the positive (top) and negative (bottom) methylation statuses; (B) the sequence of the analyzed DAT1 promoter. Numbers were assigned to individual sites in the studied region starting from 50. The methylation status of individual CpG sites was detected with a cut-o level at 20% of the G/A G ratio using 4Peaks software (Mek & Tosj, Amsterdam, The Netherlands)
Assessment of the ability to bind transcription factors
To analyze transcription binding sites for the DAT1 promoter region, PROMO software was used (http://alggen.lsi.upc.es/cgibin/promo_v3/promo/promoinit.cgi?dirDB=TF_8.3) In PROMO, for the identification of potential binding sites in sequences, weight matrices were constructed from known binding sites extracted from version 8.3 of the TRANSFAC database (http://genexplain.com/transfac/#section0) The ability of transcription factors to bind individual regions was assessed with different similarity rates i.e. 100%, 95% or 85%.
Results
We analyzed the influence of regular training on DAT1 promoter methylation status in peripheral blood leucocytes of subjects in a side-by-side comparison with healthy individuals. We observed that the level of general methylation of the CpG island was similar for both groups. Further exploration of individual CpG sites allowed to notice that there were significant differences in methylation status in positions shown in Table 1. Nonetheless, there was no rule that would indicate either higher or lower methylation of individual sites, four of them were methylated at a higher level (positions 1, 4, 5, 7, 8, 9, 10, 11, 12, 13, 16, 17, 18, 23, 25, 26, 27, 29 and 30), while one showed an inverse trend (position 3). More precise analysis with the use of the Bonferroni correction for multiple tests indicated that the CpG site methylation was higher mainly in positions 1, 4, 5, 7, 8, 9, 10, 11, 12, 13, 16, 17, 18, 23, 25, 26, 27, 29 and 30 and lower in position 3.
Table 1
Methylation status of 33 DAT1 CpG sites in sports and control group. A group of 100 athletes and 239 control individuals were studied to compare the methylation status in indicated CpG sites. The Chi square test and Spearman correlation were used. χ2(p), Chi square; OR, odds ratio; CI, Confidence Interval; R(p), Spearman correlation (-95%,+95%). * Bonferroni correction was applied to get the Bonferroni critical value (p=0.0015)
Discussion
In the current study, we analyzed 33 CpG sites of the promotor region of the DAT1 gene among a sample group of athletes and a control group. The obtained data showed significant differences between the two groups as methylation changes across all sites differed. When comparing the control and the group of athletes, the latter were hypermethylated and some were hypomethylated. What is more, the ability of transcription factors to bind to chosen sites revealed a great number of those regulators of gene expression.
One of the analyzed sites was found to be possible PAX5 transcription factor binding sites (position 3), in which the hypomethylation occurs. This is of great importance, as PAX5 is a transcription factor connected with many processes influencing the development of the nervous system. However, because of the shortage of analysis of DAT1 gene expression in our study group, we could not anticipate the precise role of this finding in connection with dopaminergic transmission. We also observed that both groups represented a similar general methylation level of the CpG islands. Interesting is the fact that more precise exploration of individual CpG sites status allowed to conclude about significant methylation status differences in positions shown in Table 1. Hypermethylation is evident in site 1 (74 vs. 40%), site 4 and 5, 7-13, 1618, 23, 25-27, 29, 30 in the research group.
It is difficult to address the results of methylation analysis in the promoter of the DAT1 gene among athletes, because of the fact that such analysis has not been conducted in any competitive groups of athletes. Similar research was conducted by Grzywacz at al. (2000) and indicated changes in island sites in the promoter of the dopamine transporter gene among cannabinoid dependent individuals. However, hypermethylation was observed only in three islands (positions 1, 6 and 28), and there was no association between endophynotype, disease entity, and hypo- or hypermethylation in the promoter of the DAT1 gene. When observing the results obtained for the group of athletes we showed a tendency in the direction of hypermethylation in the research group. However, although we should be cautious with making any conclusion, we did observe decisive hypermethylation, which can be a promising condition for further research in that area and confirmation of the chosen direction connected with the area of the dopamine transporter gene.
However, we need further studies of animal models with the usage of DAT1 knock-out animals or chromatin immunoprecipitation (ChiP) experiments which should be conducted to show which TF or other protein may bind DNA in the DAT1 promoter region. However, it could be difficult to achieve the expected results, as neural development and functionality are changing during the whole lifetime of an individual. Among probable TFs which may possibly bind the DAT1 promoted in chosen sites, SP1 can be found. SP1 is a transcription factor of low specificity that can bind a wide spectrum of target sequences.